22. Juni 2021
Regulatory Update 06/2021
Liebe Leserinnen und Leser,
die UDI-Welt dreht sich weiter, und mit ihr kommen immer mehr Aufsichtsbehörden mit ihren eigenen Anforderungen an die UDI-Konformität.
Sicherlich verfolgen Sie regelmäßig die Nachrichten über die Anforderungen, die an Sie als Medizinprodukteunternehmen und Ihren Bereich gestellt werden. Dabei müssen Sie viel Zeit aufwenden, um sich über alle relevanten Themen zu informieren.
Mit unserem Regulatory Update möchten wir Ihnen die Möglichkeit geben, sich in kurzer Zeit einen Überblick über alle UDI-relevanten Themen zu verschaffen und daraus weitere Schritte für sich abzuleiten.
Wenn Sie einen komprimierten Kurzüberblick über die relevanten Themen benötigen, scrollen Sie bitte nach unten zu tl;dr.
Inhalt
UDI in Europa
- Geräte- und Zertifikatsmodul verschoben
- Playground für M2M-Datenaustausch
- EMDN-Codes veröffentlicht
- Master UDI-DI für die Umsetzung der UDI-Anforderungen
- UDI Helpdesk
- Entwurf der Durchführungsverordnung veröffentlicht
UDI worldwide
- Südkorea I MFDS (IMDIS)
- China I NMPA (CGUDID)
- Saudi Arabien I SFDA (SAUDI-DI)
- Vereinigtes Königreich I MHRA
- Singapur I HSA (SMDR)
- Australien I TGA (AusUDID)
- Brasilien I ANVISA
- Taiwan I TFDA
Zusammenfassung und Ausblick
tl;dr
UDI in Europa
As the introduction of EUDAMED and related modules is currently a major topic, we would like to start with giving you an overview of the current situation in Europe. The most important change regarding the EU is that the MDR is applicable since May 26, 2021.
Geräte- und Zertifikatsmodul verschoben
Im März gab die Europäische Kommission bekannt, dass die Verfügbarkeit der MVP-Version (Minimum Viable Product) des zweiten Moduls, des Moduls UDI / Produktregistrierung, nun für September 2021 statt für Mai 2021 geplant ist. Dies gilt auch für das Modul zu Zertifikaten und benannten Stellen (drittes Modul). Bitte beachten Sie, dass die Eingabe von Produkt- und Zertifikatsdaten auf freiwilliger Basis erfolgt, bis EUDAMED voll funktionsfähig ist.
Die übrigen Module werden angezeigt, sobald sie danach funktionsfähig sind. Im Moment ist das einzige verfügbare Modul das Modul Akteursregistrierung, das am 1. Dezember 2020 zur Verfügung gestellt wurde. Das Modul Akteursregistrierung ist das erste von sechs Modulen.
Welche Auswirkungen hat dies auf Ihre eigene UDI-Herausforderung?
Sie sollten weiter an Ihren Projekten arbeiten und die entsprechenden Produktdaten vorbereiten, da im April bereits weitere Testphasen stattgefunden haben und die nächste im August 2021 folgen wird. (Darüber hinaus ist es wichtig, ein klares Konzept für die Basis-UDI und die Gerätezuordnung zu haben, da dies die Grundlage für Ihre MDR- und IVDR-Zertifizierungsprozesse bildet).
Playground für M2M-Datenaustausch
Wir von p36 waren im April an der neuesten Version des EUDAMED-Playgrounds für den M2M-Datenaustausch beteiligt. Gemeinsam mit unseren Kunden arbeiten wir derzeit an der Anpassung und dem Testen der neuesten Änderungen an EUDAMED, um für den voraussichtlichen Go-Live des EUDAMED UDI / Device Registration Moduls im September 2021 vorbereitet zu sein.
Diese Spielwiese ermöglicht es uns und unseren Kunden, einen Einblick in die Arbeit mit UDI-bezogenen Stammdaten innerhalb der EUDAMED zu erhalten und darauf aufbauend notwendige Anpassungen hinsichtlich der Datenverwaltung und -übermittlung vorzunehmen. Ein weiterer Playground mit erweiterten M2M-Funktionen ist für August geplant – natürlich werden wir bei den Tests dabei sein.
EMDN-Codes veröffentlicht
Die Europäische Kommission (EK) hat kürzlich eine englische Version der European Medical Device Nomenclature (EMDN) veröffentlicht. Diese Version soll von der EK genutzt werden, um Rückmeldungen zu möglichen Fehlern wie Übersetzungsfehlern oder Syntaxvorschlägen zu sammeln, um eine weitere überarbeitete Version zu entwickeln, die voraussichtlich im dritten Quartal 2021 verfügbar sein wird.
Darüber hinaus werden in der zweiten Version neue Begriffe und Beschreibungen für Medizinproduktesoftware eingeführt. Nutzer können ihre Feststellungen bis zum 4. Juni 2021 melden.
Master UDI-DI für die Umsetzung der UDI-Anforderungen
Am 4. Mai 2021 bestätigten die Europäische Kommission (EC) und die European Medical Devices Coordination Group (MDCG) die Einführung einer Master-UDI-DI zur Umsetzung der UDI-Anforderungen, basierend auf der EU MDR. Mit dieser Entscheidung reagiert die EU-Kommission auf das Bedürfnis der Unternehmen nach einer Lösung für die Zuordnung von Produkten zueinander, die stark individualisiert werden kann und so die Menge der in die EUDAMED-Datenbank einzugebenden Daten reduziert.
Zu den betroffenen Geräten gehören zum Beispiel Kontaktlinsen, Brillengläser und Lesegeräte sowie andere Produkte mit einem hohen Individualisierungsgrad, die noch nicht definiert sind. Diese Produkte müssen neue Anforderungen erfüllen, wie z.B. die folgenden:
UDI-DI (GTIN) + UDI-PIs (z. B. Chargennummer, Verfallsdatum) + Master-UDI-DI als PI + Zusätzliche neue PIs (z. B. klinische Größe, verwendetes Material, Durchmesser, optische Stärke).
Welche Fristen muss die Industrie beachten?
- Nach derzeitigem, unbestätigtem Stand müssen die Vergabestellen bis spätestens Ende September 2021 (muss noch bestätigt werden) relevante Normen für die Master-UDI-DI als PI und für zusätzliche neue PIs (d.h. klinische Größe, verwendetes Material, Durchmesser, optische Dicke) bereitstellen.
- Die Vergabe von Basic UDI-DI, UDI-DI und UDI-PI läuft bis zum 26. Mai 2021. Um die Flexibilität der Medizintechnikunternehmen bis zur Bereitstellung der Master-UDI-DI zu gewährleisten, entwickelt die EK derzeit Übergangsmaßnahmen.
- Die Kennzeichnung von Produkten wird je nach Risikoklasse zwischen 2023 und 2025 verpflichtend werden. Die Produkte können dann mit der Master-UDI-DI gekennzeichnet werden.
- Die Registrierung von Produkten in der EUDAMED-Datenbank, einschließlich der Master-UDI-DI, wird ab 2024 möglich sein. Eine frühere freiwillige Registrierung ist nicht möglich.
Aufgrund der vielfältigen Themen steht Europa im Hinblick auf die Einführung der EUDAMED vor einem großen Wandel im Bereich der UDI. Dies ist nicht nur in Europa, sondern weltweit zu beobachten, wie der folgende Abschnitt verdeutlicht.
UDI Helpdesk
Die Europäische Kommission stellt einen Helpdesk zur Verfügung, der die Hersteller von Medizinprodukten bei der Umsetzung der entsprechenden Anforderungen unterstützt. Neben der UDI-Vergabe, Kennzeichnung und Registrierung von Produkten in Europa bietet der Helpdesk auch Unterstützung bei der Verwendung der Europäischen Nomenklatur für Medizinprodukte (EMDN), die die EK den betroffenen Unternehmen zur Verfügung gestellt hat.
Entwurf der Durchführungsverordnung veröffentlicht
Die Europäische Kommission hat den Entwurf des Durchführungsrechtsakts zu EUDAMED für eine öffentliche Konsultation freigegeben. Vom 25. Mai 2021 bis zum 22. Juni 2021 hat die Industrie die Möglichkeit, den Entwurf zu kommentieren und Feedback zu geben. Die Rückmeldungen werden von der Europäischen Kommission berücksichtigt, um diese Initiative fertig zu stellen.
UDI weltweit
Neben Europa erhöhen auch andere Behörden das Tempo ihrer eigenen Anforderungen. Der folgende Abschnitt gibt daher einen kurzen Überblick über die unserer Meinung nach wichtigsten bevorstehenden Änderungen für Hersteller von Medizinprodukten weltweit.
Südkorea I MFDS (IMDIS)
Die Geräte der Klasse III sind seit dem 1. Juli 2020 verfügbar und die nächsten Geräte der Klasse II werden am 1. Juli 2021 verfügbar sein. In Zusammenarbeit mit unseren Kunden haben wir das Content Package für Südkorea neu aufgelegt, einschließlich einer Machine-2-Machine-Schnittstelle, um UDI-Daten direkt von unserer UDI Platform in die südkoreanische IMDIS-Datenbank registrieren zu können.
China I NMPA (CGUDID)
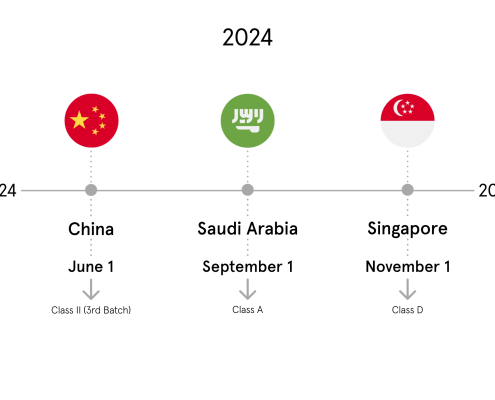
Seit dem 1. Januar 2021 müssen Hochrisikoprodukte der Klasse III, z. B. Herz- oder Hirnimplantate/Prothesen, in der chinesischen UDI-Datenbank CGUDID registriert werden. Ab dem Umsetzungsdatum müssen alle Medizinprodukte, die in den Anwendungsbereich dieser Produkttypen fallen und ab diesem Datum hergestellt werden, mit einer UDI-Kennzeichnung versehen sein.
Der nächste Schritt für die Registrierung von Medizinprodukten in China wird folgender sein:
- Abschluss der Einreichung der restlichen Produkte der Klasse III bis zum 1. Januar 2022.
- Einreichung der UDI-bezogenen Daten für Produkte der Klasse II bis zum 1. August 2024.
- Als letzter Schritt müssen alle verbleibenden Produkte bis zum 1. August 2026 bei der CGUDID eingereicht werden.
Unsere Freigabe für das China NMPA Content Package ist für die zweite Hälfte des Jahres 2021 geplant.
Saudi Arabien I SFDA (SAUDI-DI)
Die Verfügbarkeit von Geräten der Klasse D in Saudi-Arabien ist auf den 1. August 2021 festgelegt. Daher ist die Veröffentlichung des entsprechenden Content Package für die zweite Hälfte des Jahres 2021 geplant.
Bislang bietet die SFDA keinen Datentransfer oder Massenupload über Excel-Sheets an, so dass Medizinproduktehersteller keine M-2-M Schnittstelle zur Einreichung nutzen können, sondern alle relevanten Daten selbst in die Datenbank (Saudi-DI) eingeben müssen. Allerdings orientiert sich die SFDA stark an den IMDFR-Spezifikationen (International Medical Device Regulators Forum), was einen großen Vorteil für die Unternehmen bietet, da sich ca. 80% der Daten aus bereits vorhandenen Daten zusammensetzen, die bei FDA und EUDAMED eingereicht werden müssen. Folglich müssen nur etwa 20 % der erforderlichen Daten für die Registrierung neuer Produkte gesondert vorgelegt werden.
Die SFDA arbeitet an der Bereitstellung eines Excel-basierten Daten-Uploads. Hier können wir unterstützen, indem wir die in der UDI Platform gepflegten Daten als Excel-basierten Download zur Verfügung stellen, der zum Import der Daten in die SAUDI-DI Datenbank genutzt werden kann.
Vereinigtes Königreich I MHRA
Da immer mehr Länder ihre eigenen regulatorischen Anforderungen veröffentlichen, wurden wir von mehreren Medizinprodukteherstellern um weitere Informationen zu den Anforderungen in Großbritannien gebeten.
Seit dem 1. Januar 2021 gibt es mehrere Änderungen für die Platzierung von Produkten in Großbritannien (beinhaltet England, Wales und Schottland):
- Die CE-Kennzeichnung wird in Großbritannien noch bis zum 30. Juni 2023 anerkannt.
- Zertifikate, die von EU-anerkannten benannten Stellen ausgestellt werden, sind bis zum 30. Juni 2023 weiterhin für den britischen Markt gültig.
- Die EU erkennt die benannten Stellen des Vereinigten Königreichs nicht mehr an, und die benannten Stellen des Vereinigten Königreichs, können keine CE-Zertifikate mehr ausstellen.
- Hersteller von Produkten der Klasse I, Sonderanfertigungen und allgemeinen IVDs, die ihre Produkte bereits vor dem 1. Januar 2021 bei der MHRA registrieren lassen mussten, müssen ihre Produkte ab dem 1. Januar 2021 auf derselben Grundlage registrieren lassen, da die oben genannten Registrierungsfristen für diese Produkte nicht gelten.
- Da die in der EU-MDR und der EU-IVDR enthaltenen Bestimmungen in Großbritannien nicht in nationales Recht umgesetzt werden, werden sie auch nicht in Großbritannien eingeführt.
- Hersteller mit Sitz außerhalb Großbritanniens, die ein Produkt in Großbritannien in Verkehr bringen wollen, müssen eine einzige verantwortliche Person in Großbritannien benennen, die die Verantwortung für das Produkt in Großbritannien übernimmt. Diese verantwortliche Person im Vereinigten Königreich muss einen eingetragenen Geschäftssitz im Vereinigten Königreich haben.
Die folgenden Produkte müssen bei der MHRA registriert werden, wenn sie ab dem 1. Mai 2021 in Großbritannien auf den Markt gebracht werden sollen:
- aktive implantierbare Medizinprodukte
- Medizinprodukte der Klasse III
- Implantierbare Medizinprodukte der Klasse IIb
- IVD-Produkte der Liste A
Die folgenden Produkte müssen bei der MHRA registriert werden, wenn sie ab dem 1. September 2021 in Großbritannien auf den Markt gebracht werden sollen:
- Nichtimplantierbare Medizinprodukte der Klasse IIb
- Medizinprodukte der Klasse IIa
- Produkte der IVD-Liste B
- IVDs zur Eigenanwendung
Die folgenden Produkte müssen bei der MHRA registriert werden, wenn sie ab dem 1. Januar 2022 in Großbritannien auf den Markt gebracht werden sollen:
- Medizinprodukte der Klasse I
- allgemeine IVDs
Es ist möglich, Produkte vor den oben genannten Terminen zu registrieren, aber es besteht keine rechtliche Verpflichtung dazu.
Es ist möglich, Produkte vor den oben genannten Terminen zu registrieren, aber es besteht keine rechtliche Verpflichtung dazu.
Hersteller von Produkten der Klasse I, Sonderanfertigungen und allgemeinen IVD, die vor dem 1. Januar 2021 verpflichtet waren, ihre Produkte bei der MHRA zu registrieren (d. h. wenn der Hersteller seinen Sitz im Vereinigten Königreich oder der Bevollmächtigte seinen Sitz in Nordirland hat), müssen ihre Produkte weiterhin auf derselben Grundlage wie bisher registrieren, da die oben genannten Registrierungsfristen für diese Produkte nicht gelten. Wenn ein Medizinprodukt bereits vor dem 1. Januar 2021 bei der MHRA registriert war, muss es nicht erneut registriert werden.
Die Hersteller von Medizinprodukten müssen der MHRA über das Online-Registrierungssystem für Medizinprodukte (MHRA DORS) Informationen über das Unternehmen, das Medizinprodukt und die verantwortliche Person im Vereinigten Königreich (UKRP) vor Ablauf der jeweiligen Registrierungsfristen übermitteln. Die Unternehmen müssen ein MHRA DORS-Konto einrichten, bevor sie ihre Registrierungsdaten in das Online-System eingeben.
Singapur I HSA (SMDR)
Am 25. Mai 2021 veröffentlichte Singapur den Entwurf der „Guidance on the Medical Device Unique Device Identification (UDI) System“ (Leitfaden zum System zur eindeutigen Identifizierung von Medizinprodukten (UDI)), in dem dargelegt wird, wie das Land die Verwaltung und Einreichung von UDI-bezogenen Daten handhaben will.
Darüber hinaus kündigte Singapur an, dass es ein UDI-System einführen wird, dessen Schlüsselelemente auf den international harmonisierten Grundsätzen des International Medical Device Regulators Forum (IMDRF) basieren.
UDIs, die auf den Etiketten von Medizinprodukten für die Märkte in der EU oder den USA angebracht sind, werden in Singapur unverändert akzeptiert.
Die UDI-bezogenen Datenelemente für alle Risikoklassen von Medizinprodukten können auf freiwilliger Basis aktualisiert werden, noch bevor der Stichtag für die jeweilige Umsetzungsphase in Kraft tritt. Diese Konformitätsfristen helfen den Herstellern von Medizinprodukten bei der Planung, wann sie ihre Medizinprodukte bei der SMDR registrieren müssen, falls sie diese in Singapur vertreiben wollen:
- Phase 1 mit allen Koronarstents, orthopädischen Gelenkersatzimplantaten und Intraokularlinsen beginnt im November 2022
- Phase 2, die alle allgemeinen Medizinprodukte der Klasse D und IVDs umfasst, gilt ab dem 1. November 2024,
- Phase 3, die alle allgemeinen Medizinprodukte der Klasse C und IVDs umfasst, beginnt am 1. November 2026,
- Phase 4 mit allen allgemeinen Medizinprodukten der Klasse B und IVDs beginnt am 1. November 2028.
- Alle UDIs von allgemeinen Medizinprodukten der Klasse A und IVDs können auf freiwilliger Basis eingeführt werden.
Nach Abschluss der ersten Phase wird diese überprüft und die Erfahrungen werden genutzt, um die Datenbank und die Anforderungen für die folgenden Implementierungsphasen in SMDR neu zu justieren.
Australien I TGA (AusUDID)
Die TGA wird ihre eigene UDI-Datenbank, die Australian Unique Device Identification Database (AusUDID), einrichten und pflegen. Ein offizieller Leitfaden soll in naher Zukunft weitere Einzelheiten zur Umsetzung der UDI-Anforderungen enthalten.
Derzeit werden Optionen für die Einführung eines australischen UDI-Systems (Unique Device Identification) erkundet. Nach derzeitigem Stand wird das australische UDI-System auf dem UDI-Leitfaden der IMDRF basieren, wobei die Regeln und Richtlinien auf denen der Vergabestellen in der EU und den USA beruhen.
Brasilien I ANVISA
In den letzten Monaten hat die ANVISA als zuständige Behörde für Medizinprodukte in Brasilien an einem RDC-Entwurf (Resolution of the Board of Directors) für die Registrierung von Medizinprodukten in Brasilien gearbeitet.
Nach der Vorlage dieses Entwurfs zur Prüfung bei der ANVISA im April und der Analyse der regulatorischen Auswirkungen, die von Mai bis Juli 2021 stattfinden wird, wird die Öffentlichkeit zu ihrer Meinung über die UDI RDC befragt werden. Derzeit ist geplant, dass die Bewertung und Veröffentlichung der RDC zu UDI durch das Dicol (Board of Directors) Ende dieses Jahres erfolgen wird.
Taiwan I TFDA
Neben den bereits geltenden UDI-Gesetzen und bevorstehenden zwingenden Ereignissen wie der Klasse-D-Frist in Saudi-Arabien hat Taiwan gerade sein eigenes UDI-Anforderungspapier veröffentlicht. Wir sehen eine Adaption der IMDRF-Richtlinie und werden rechtzeitig ein Content Package für die UDI Platform vorlegen.
Zusammenfassung und Ausblick
Die Fülle der Themen und die Vielfalt der Anforderungen der verschiedenen Länder zeigen, dass es immer wieder zu Änderungen der relevanten Vorschriften/Gesetzgebungen kommt. Medizinproduktehersteller müssen sich, je nach Markt, in dem sie tätig sind, damit auseinandersetzen und einen Umsetzungsplan in ihrem Unternehmen erstellen.
Diejenigen unter Ihnen, die langfristig planen und sich frühzeitig global aufstellen, können bereits erste Synergien aus der Nutzung der UDI Platform nutzen, denn die Plattform nutzt das einzigartige Common Device Data Model, wodurch ein großer Teil der Daten aus bestehenden FDA- und EUDAMED-Daten wiederverwendet werden kann. Dies bietet Ihnen als MedTech-Unternehmen den großen Vorteil, dass Sie sich auf die marktspezifischen Besonderheiten konzentrieren können und dabei nicht nur Ihre Ressourcen, sondern auch Zeit sparen und UDI-bezogene Prozesse effektiver gestalten können.
Die so gewonnene Zeit können Sie nutzen, um sich auf Ihr Kerngeschäft zu konzentrieren und Ihre langfristige Wettbewerbsfähigkeit zu sichern.
Falls Sie weitere detaillierte Informationen zu diesen Themen benötigen oder nach einer Anleitung suchen, wie Sie mit dieser zunehmenden globalen Herausforderung umgehen können, zögern Sie bitte nicht, uns zu kontaktieren unter info@p36.io.
tl;dr
UDI in Europa
- Die EU-MDR ist seit dem 26. Mai 2021 anwendbar.
- Die Verfügbarkeit der MVP-Version (Minimum Viable Product) des zweiten Moduls, des Moduls UDI/Geräteregistrierung und des Moduls zu Zertifikaten und benannten Stellen (drittes Modul) ist nun für September 2021 statt Mai 2021 geplant.
- Wir von p36 waren im April an der neuesten Version des EUDAMED-Playgrounds für den M2M-Datenaustausch beteiligt und werden am nächsten Test teilnehmen, der für August geplant ist, um Einblicke in die Arbeit mit UDI-bezogenen Stammdaten innerhalb von EUDAMED zu erhalten.
- Die Europäische Kommission hat eine englische Version der Europäischen Nomenklatur für Medizinprodukte (EMDN) veröffentlicht, um Rückmeldungen zu möglichen Fehlern einzuholen, damit eine weitere überarbeitete Version entwickelt werden kann, die voraussichtlich im dritten Quartal 2021 verfügbar sein wird.
- Sowohl die EK als auch die MDCG bestätigten die Einführung einer Master-UDI-DI für die Umsetzung der UDI-Anforderungen, die auf der EU-MDR basiert. Mit dieser Entscheidung reagiert die EK auf das Bedürfnis der Unternehmen nach einer Lösung, um Produkte einander zuzuordnen, die stark individualisiert sein können (z.B. Kontaktlinsen) und auf diese Weise die Menge der in die EUDAMED-Datenbank einzugebenden Daten zu reduzieren.
- Seit dem 26. Mai 2021 stellt die Europäische Kommission einen Helpdesk zur Verfügung, der die Hersteller von Medizinprodukten bei der Umsetzung der entsprechenden Anforderungen unterstützt. Sie bietet auch Unterstützung bei der Nutzung des EMDN, das die EK den betroffenen Unternehmen zur Verfügung gestellt hat.
- Die EK hat den Entwurf des Durchführungsrechtsakts zu EUDAMED für eine öffentliche Konsultation freigegeben. Bis zum 22. Juni 2021 hat die Industrie die Möglichkeit, diesen Entwurf zu kommentieren und Rückmeldungen zu geben.
UDI weltweit
- Südkorea I Die Geräte der Klasse II werden am 1. Juli 2021 verfügbar sein Wir haben das Content Package für Südkorea freigegeben, das eine M-2-M-Schnittstelle enthält, um UDI-Daten direkt von unserer UDI Platform in die südkoreanische IMDIS-Datenbank registrieren zu können.
- China I Seit dem 1. Januar 2021 müssen Hochrisikoprodukte der Klasse III, z.B. Herz- oder Hirnimplantate/Prothesen, in der chinesischen UDI-Datenbank CGUDID registriert werden. Unsere Freigabe für das China NMPA Content Package ist für die zweite Hälfte des Jahres 2021 geplant.
- Saudi-Arabien I Die Anwendung von Produkten der Klasse D in Saudi-Arabien ist auf den 1. August 2021 festgelegt. Daher ist die Veröffentlichung des entsprechenden Content Package für die zweite Jahreshälfte 2021 geplant. Derzeit bietet die SFDA keinen Datentransfer oder Massen-Upload über Excel-Sheets an, arbeitet aber derzeit an einem Excel-basierten Daten-Upload.
- UK I Ab dem 1. Mai 2021 müssen aktive implantierbare Medizinprodukte, Medizinprodukte der Klasse III, implantierbare Medizinprodukte der Klasse IIb und Produkte der IVD-Liste A bei der MHRA registriert werden, wenn Medizinproduktehersteller ihr Produkt in Großbritannien auf den Markt bringen wollen. Der nächste Termin für die Einhaltung der Vorschriften ist der 1. September 2021.
- Singapur I Kürzlich hat Singapur den Entwurf eines Leitfadens zum UDI-System (Medical Device Unique Device Identification)“ veröffentlicht, in dem es darlegt, wie es mit UDI-bezogenen Daten umgehen will. Sie werden ein UDI-System einführen, dessen Schlüsselelemente auf den vom IMDRF veröffentlichten Grundsätzen beruhen. UDIs, die auf den Etiketten von Medizinprodukten für die Märkte in der EU oder den USA angebracht sind, werden in Singapur unverändert akzeptiert. Phase 1 beginnt im November 2022.
- Australien I Die TGA wird eine eigene UDI-Datenbank, die Australian Unique Device Identification Database (AusUDID), einrichten und pflegen. Ein offizieller Leitfaden soll in naher Zukunft weitere Einzelheiten zur Umsetzung der UDI-Anforderungen enthalten.
- Brasilien I ANVISA, die zuständige Behörde für Medizinprodukte in Brasilien, hat an einem RDC-Entwurf für die Registrierung von Medizinprodukten in Brasilien gearbeitet. Derzeit ist geplant, dass die Bewertung und Veröffentlichung der RDC zu UDI durch Dicol (Board of Directors) Ende dieses Jahres erfolgt.
- Taiwan I Taiwan hat gerade sein eigenes UDI-Anforderungspapier veröffentlicht, in dem wir eine Adaption der IMDRF-Richtlinie sehen. Wir werden zu gegebener Zeit ein Content Package für die UDI Platform vorlegen.