June 22, 2021
Regulatory Update 06/2021
Dear reader,
the UDI world is spinning around and along with it more and more regulatory agencies are coming along with their own requirements for UDI compliance.
Certainly, you monitor the news about the requirements that are placed on you as a medical device company and your area on a regular basis. In doing so, you have to spend a lot of time to inform yourself about all relevant topics.
With our regulatory update, we would like to provide you with the opportunity of obtaining an overview of all UDI-related relevant topics in a short time and deriving further steps for yourself.
If you require a compressed short overview about the relevant topics, please scroll down to tl;dr.
Content
UDI in Europe
- Device and certificate module postponed
- Playground for M2M data exchange
- EMDN codes published
- Master UDI-DI for implementation of UDI requirements
- UDI Helpdesk
- Draft Implementing act released
UDI worldwide
- South Korea I MFDS (IMDIS)
- China I NMPA (CGUDID)
- Saudi Arabia I SFDA (SAUDI-DI)
- UK I MHRA
- Singapore I HSA (SMDR)
- Australia I TGA (AusUDID)
- Brazil I ANVISA
- Taiwan I TFDA
Conclusion and outlook
tl;dr
UDI in Europe
As the introduction of EUDAMED and related modules is currently a major topic, we would like to start with giving you an overview of the current situation in Europe. The most important change regarding the EU is that the MDR is applicable since May 26, 2021.
Device and certificate module postponed
In March, the European Commission announced that the availability of the MVP (Minimum Viable Product) version of the second module, the UDI / device registration module, is now planned for September 2021 instead of May 2021. This also applies to the module on Certificates and Notified Bodies (third module). Please note that entering device and certificate data will be on voluntary basis until EUDAMED is fully operational.
The remaining modules will be displayed as soon as they are functional afterwards. As of now, the only available module is the Actor registration module, which has been made available on 1st December 2020. The Actor registration module is the first of six modules.
Which impact does this have on your own UDI challenge?
You should keep working on your projects and prepare the relevant product data, as further testing phases already took place in April and the next one is about to follow in August 2021. (In addition, it is important to have a clear concept for Basic UDI and device assignment as this forms the basis for your MDR and IVDR certification processes).
Playground for M2M data exchange
We from p36 were involved in the newest version of the EUDAMED playground for M2M data exchange in April. Together with our customers we are currently working on the adaption and testing of the latest changes to EUDAMED to be prepared for the anticipated go live of the EUDAMED UDI / device registration module in September 2021.
This playground enables us and our customers to get an insight in working with UDI-related master data within the EUDAMED and based on this, make necessary adaptions regarding data governance and submission. Another playground with extended M2M capabilities is planned for August – of course we will be part of the testing.
EMDN codes published
The European Commission (EC) recently published an English version of the European Medical Device Nomenclature (EMDN). This version is intended to be used by the EC to gather feedback on possible errors, such as translation mistakes or syntax suggestions, in order to develop a further revised version, which is expected to be available in the third quarter of 2021.
Apart from this, new terms and descriptions for medical device software will be rolled out in the second version. Users can report their findings until June 4, 2021.
Master UDI-DI for implementation of UDI requirements
On May 4, 2021 the European Commission (EC) and the European Medical Devices Coordination Group (MDCG) confirmed the introduction of a Master UDI-DI for the implementation of UDI requirements, based on the EU MDR. With this decision, the EC is responding to the need of companies for a solution to assign devices to each other, which can be highly individualized and in this way reduce the volume of data to be entered into the EUDAMED database.
The affected devices include, for example, contact lenses, spectacle lenses and ready readers, as well as other products with a high level of individualization, which are not yet defined. These devices have to comply with new requirements, such as the following:
UDI-DI (GTIN) + UDI-PIs (e.g. lot number, expiry date) + Master UDI-DI as a PI + Additional new PIs (e.g. clinical size, material used, diameter, optical strength).
The Master UDI-DI will be captured in a UDI barcode as PI (Product Identifier), which will be placed on the packaging/label of the product. The specific use as well as the length, structure, etc. of the Master UDI-DI is not known yet and therefore still has to be defined by EUDAMED. Therefore, the accredited issuing agencies have to define how the Master UDI-DI should be displayed. However, the aim is to register the Master UDI-DI instead of the UDI-DI.
Which timelines does the industry have to keep in mind?
- According to current unconfirmed status, Issuing Entities must provide relevant standards for the Master UDI-DI as PI and for additional new PIs (i.e., clinical size, material used, diameter, optical thickness) at least by the end of September 2021 (has to be confirmed).
- The assignment of Basic UDI-DI, UDI-DI and UDI-PI will run until May 26, 2021. In order to ensure the flexibility of medical technology companies until the Master UDI-DI is made available, the EC is currently developing transitional measures.
- The labeling of devices will become mandatory between 2023 and 2025, depending on the respective risk class. Products can then be labeled including the Master UDI-DI.
- The registration of devices in the EUDAMED database, including the Master UDI-DI, will be possible in 2024. Earlier voluntary registration will not be allowed.
As a result of the multiple topics, Europe is facing a major transformation in the field of UDI with regard to the introduction of EUDAMED. This can be observed not only in Europe, but worldwide, as the following section will illustrate.
UDI Helpdesk
The European Commission provides a helpdesk to assist medical device manufactureres in implementing the relevant requirements. In addition to the UDI assignment, labeling and registration of devices in Europe, the helpdesk also provides support in the use of the European Medical Devices Nomenclature (EMDN), which the EC made available to the affected companies.
Draft Implementing act released
The EC released the draft Implementing act on EUDAMED for a public consultation. From May 25, 2021 up to June 22, 2021 the industry has the opportunity to comment this draft by giving feedback. The feedback will be taken by the EC to finalize this initiative.
UDI worldwide
In addition to Europe, other authorities are increasing speed with their own requirements. The following section therefore provides a brief overview of what we consider to be the most important upcoming changes for medical device manufacturers worldwide.
South Korea I MFDS (IMDIS)
The Class III devices are applicable since July 1, 2020 and the next Class devices from Class II will be applicable on July 1, 2021. In collaboration with our customers, we have relased the Content Package for South Korea, including a Machine-2-Machine interface in order to be able to register UDI-data directly from our UDI Platform to the South Korean IMDIS-database.
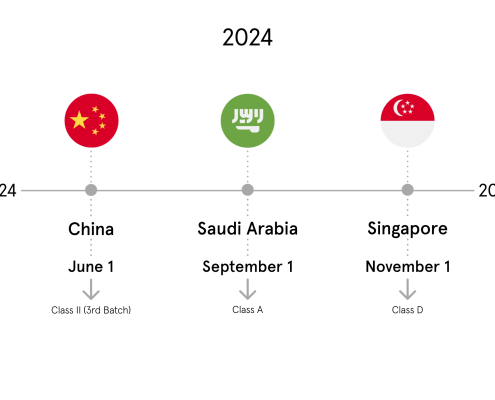
China I NMPA (CGUDID)
Since January 1, 2021 high-risk Class III devices, e.g. heart or brain implants/prostheses, must be registered in the Chinese UDI-database CGUDID. From the Implementation Date, all medical devices falling within the scope of these device types, which are manufactured from this date onwards, will be required to possess a UDI marking.
The next step for registering medical devices in China will be the following:
- Completing of the submission of the remaining devices from Class III until January 1, 2022.
- Submission of the UDI-rleated data for Class II devices until August 1, 2024.
- As the final step, all remaining devices have to be submitted to CGUDID until August 1, 2026.
Our release for the China NMPA Content Package is planned for the second half of 2021.
Saudi Arabia I SFDA (SAUDI-DI)
The application of Class D devices in Saudi Arabia is set to August 1, 2021. Therefore our release of the referring Content Package is planned for the second half of 2021.
As of now, the SFDA does not offer any data transfer or mass upload via excel sheets, so medical device manufacturers can’t use any M-2-M interface for submission, but instead of this have to type in all relevant data by themselves into the database (Saudi-DI). However, the SFDA is strongly oriented to the IMDFR (International Medical Device Regulators Forum) specifications, which offers a great benefit for companies, as about 80% of the data is composed of already existing data that has to be submitted to FDA and EUDAMED. Consequently, only about 20% of the required data have to be provided separately in order to register new devices.
The SFDA is working on providing an Excel-based Data upload. Here we can support with providing the data maintained in the UDI Platform as Excel-based download which can be used for importing the data into the SAUDI-DI database.
UK I MHRA
In addition to more and more countries publishing their own regulatory requirements, multiple medical device manufacturers contacted us for further information regarding the requirements of the UK.
Since January 1, 2021 there have been several changes for the placement of devices in Great Britain (contains England, Wales and Scotland):
- CE marking will continue to be recognized in Great Britain until June 30, 2023.
- Certificates that are issued by EU-recognized Notified Bodies will continue to be valid for the Great Britain market until June 30, 2023.
- The EU no longer recognizes UK Notified Bodies and UK Notified Bodies, that have become UK Approved Bodies, are not able to issue CE certificates.
- Manufacturers of Class I devices, custom-made devices and general IVDs that, prior to January 1, 2021, already had to register their devices with the MHRA are required to register their devices from January 1, 2021 on the same basis, as the above registration deadlines will not apply to those devices.
- As the provisions contained within the EU MDR and EU IVDR will not be transposed into law in Great Britain, the will not be implemented in Great Britain.
- Manufacturers based outside the UK, wishing to place a device on the Great Britain market, need to appoint a single UK Responsible Person to take responsibility for the product in Great Britain. This UK Responsible Person needs a registered place of business within the UK.
The following devices must be registered with the MHRA if they are being placed on the Great Britain market from May 1, 2021:
- active implantable medical devices
- Class III medical devices
- Class IIb implantable medical devices
- IVD List A products
The following devices must be registered with the MHRA if they are being placed on the Great Britain market from September 1, 2021:
- Class IIb non-implantable medical devices
- Class IIa medical devices
- IVD List B products
- self-test IVDs
The following devices must be registered with the MHRA if they are being placed on the Great Britain market from January 1, 2022:
- Class I medical devices
- general IVDs
It is possible to register devices ahead of the above dates, but there is no legal obligation to do so.
Manufacturers of Class I devices, custom-made devices and general IVDs that, prior to 1 January 2021, were required to register their devices with the MHRA (i.e. where the manufacturer is based in the UK or the Authorized Representative is based in Northern Ireland) must continue to register their devices on the same basis as they did previously as the above registration timings will not apply to these devices. Where a medical device was already registered with the MHRA before 1 January 2021, it does not need to be re-registered.
On or before their applicable registration deadlines, device manufacturers must submit company, device and UK Responsible Person (UKRP) information to MHRA via the regulator’s Device Online Registration System (MHRA DORS). Companies must set up an MHRA DORS account before entering their registration data into the online system.
Singapore I HSA (SMDR)
On May 25, 2021, Singapore published its draft “Guidance on the Medical Device Unique Device Identification (UDI) System” on how they intend to handle UDI-related data governance and submission.
In addition, Singapore announced that they will introduce a UDI-system whose key elements are based on the internationally harmonized principles published by the International Medical Device Regulators Forum (IMDRF).
UDIs placed on the labels of medical devices for the markets in the EU or USA will be accepted unchanged for Singapore.
UDI-related data elements for all medical device risk classes can be updated on a voluntary basis even before the compliance date for the respective implementation phase becomes effective. These compliance deadlines help medical device manufacturers to plan when to register their medical devices with the SMDR, in case they plan to distribute them in Singapore:
- Phase 1 with all Coronary stents, orthopaedic joint replacement implants and Intraocular lens starts in November 2022
- Phase 2, including all Class D General medical devices and IVDs will be applicable from November 1, 2024,
- Phase 3, including all Class C General medical devices and IVDs starts on November 1, 2026,
- Phase 4 with all Class B General medical devices and IVDs starts on November 1, 2028
- All UDIs of Class A General medical devices and IVDs can be introduced on a voluntary basis.
After the first phase has been completed, it will be reviewed and the experiences will be used to readjust the database and requirements for the following implementation phases into SMDR.
Australia I TGA (AusUDID)
The TGA will establish and maintain its own UDI database, called Australian Unique Device Identification Database (AusUDID). An official guide is expected to provide further details on the implementation of UDI requirements in the near future.
Currently, options for the introduction of an Australian Unique Device Identification (UDI) system are being explored. The current status is that the Australian UDI system will be based on the IMDRF UDI guidance, with rules and guidelines based on those of the issuing bodies in the EU and U.S..
Brazil I ANVISA
In the last months, ANVISA as the responsible authority for medical devices in Brazil, has been working on a RDC (Resolution of the board of directors) draft for the registration of medical devices in Brazil.
After submitting this draft for consideration to ANVISA in April and the analysis of the regulatory impact which will take place from May until July 2021, the public will be consulted regarding their opinion on UDI RDC. Currently, it is planned that the evaluation and publication of the RDC on UDI by Dicol (Board of directors) will take place at the end of this year.
Taiwan I TFDA
Next to already applicable UDI legislations and upcoming compelling events like Saudi Arabia Class D deadline, Taiwan has just published its own UDI requirements paper. We see an adaption of the IMDRF guideline and will come up with a Content Package for the UDI Platform in time.
Conclusion and outlook
The abundance of topics and the diversity of the requirements of different countries show that changes in the relevant regulations / legislations occur frequently. Medical device manufacturers, depending on the markets in which they operate, must face and create an implementation plan in their company.
Those of you who plan for the long term and position themselves globally at an early stage can already take advantage of the first synergies resulting from the use of the UDI Platform, as the platform uses the unique common device data model, whereby a huge part of the data can be reused from existing FDA and EUDAMED data. This offers you as a MedTech company the great advantage, that you can focus on the market-specific specialties and in doing so not only save your resources but also save time and make UDI-related processes more effective.
This saved time can be used to focus on your core business and to ensure your long-term competitiveness.
In case you need further detailed information on those topics or if you are searching for guidance how to deal with this increasing global challenge, please don’t hesitate to contact us at info@p36.io.
tl;dr
UDI in Europe
- The EU MDR is applicable since May 26, 2021.
- The availability of the MVP (Minimum Viable Product) version of the second module, the UDI / device registration module and the module on Certificates and Notified Bodies (third module), is now planned for September 2021 instead of May 2021.
- We from p36 were involved in the newest version of the EUDAMED playground for M2M data exchange in April and will be part of the next testing, which is planned for August, to get insights in working with UDI-related master data within EUDAMED.
- The EC published an English version of the European Medical Device Nomenclature (EMDN) to gather feedback on possible errors, in order to develop a further revised version, which is expected to be available in the third quarter of 2021.
- Both, EC and MDCG confirmed the introduction of a Master UDI-DI for the implementation of UDI requirements, based on the EU MDR. With this decision, the EC is responding to the need of companies for a solution to assign devices to each other, which can be highly individualized (e.g. contact lenses) and in this way reduce the volume of data to be entered into the EUDAMED database.
- Since May 26, 2021 the EC provided a helpdesk to assist medical device manufactureres in implementing the relevant requirements. It also provides support in the use of the EMDN, which the EC made available to the affected companies.
- The EC released the draft Implementing act on EUDAMED for a public consultation. Until June 22, 2021 the industry has the opportunity to comment this draft by giving feedback.
UDI worldwide
- South Korea I The Class II devices will be applicable on July 1, 2021. We have released the Content Package for South Korea, including a M-2-M interface in order to be able to register UDI-data directly from our UDI Platform to the South Korean IMDIS-database.
- China I Since January 1, 2021 high-risk Class III devices, e.g. heart or brain implants/prostheses, must be registered in the Chinese UDI-database CGUDID. Our release for the China NMPA Content Package is planned for the second half of 2021.
- Saudi Arabia I The application of Class D devices in Saudi Arabia is set to August 1, 2021. Therefore our release of the referring Content Package is planned for the second half of 2021. As of now, the SFDA does not offer any data transfer or mass upload via excel sheets, but is currently working on an Excel-based Data upload.
- UK I From May 1, 2021, active implantable medical devices, Class III medical devices, Class IIb implantable medical devices and IVD List A products have to be registered with the MHRA if medical device manufacturers want to place their product on the Great Britain market. The next compliance date is September 1, 2021.
- Singapore I Recently Singapore published its draft “Guidance on the Medical Device Unique Device Identification (UDI) System” on how they intend to handle UDI-related data. They will introduce a UDI-system whose key elements are based on the principles published by the IMDRF. UDIs placed on the labels of medical devices for the markets in the EU or USA will be accepted unchanged for Singapore. Phase 1 starts in November 2022.
- Australia I The TGA will establish and maintain its own UDI database, called Australian Unique Device Identification Database (AusUDID). An official guide is expected to provide further details on the implementation of UDI requirements in the near future.
- Brazil I ANVISA, the responsible authority for medical devices in Brazil, has been working on a RDC draft for the registration of medical devices in Brazil. Currently, it is planned that the evaluation and publication of the RDC on UDI by Dicol (Board of directors) will take place at the end of this year.
- Taiwan I Taiwan has just published its own UDI requirements paper where we see an adaption of the IMDRF guideline. We will come up with a Content Package for the UDI Platform in time.